

Strike True: How to accurately target your desired neuron population in tissue slices

by Martyna Panasiuk, Postdoc at the Centre of Developmental Neurobiology, King's College London
Introduction
In the low-throughput world of patch clamp electrophysiology, thoughtfully defining your cell population of interest is one of the most crucial elements of experiment design. Bad news is, most electrophysiologists’ favourite cells – neurons – show an incredibly high diversity of cell types. These types can often be split into subtypes (or subtypes of subtypes…) and vary in their properties depending on the brain area (or the sub-area of a sub-area…) where they’re present.
Additionally, working with live cells limits our options when it comes to labelling and identifying the cells of interest during the experiment. Many a PhD student has woken up in cold sweat, paranoid that their dataset of cortical layer 5a pyramidal neurons might actually be contaminated with (gasp) pyramidal neurons from layer 5b! What’s worse, unlike when working with fixed tissue samples, there can be limited opportunity to go back and check what cell type a given recording truly came from. This guide aims to minimise such PhD nightmares by giving general tips and strategies to confidently identifying and patching the cell population that you actually meant to patch.
Identifying your target brain region
Measure twice, slice once
The single most important step in being able to identify your region of interest (ROI) is becoming familiar with an appropriate reference atlas. Nowadays, digital brain atlases like the Allen Brain Atlas are increasingly popular and accessible. But keep in mind that having a physical copy of an atlas – either a full book copy, or at least a few printed pages showing your ROI – can be extremely useful while you’re actually dissecting and sectioning at the vibratome.
Ideally, your atlas should be in the same orientation as the plane of your slicing – e.g. The Allen Mouse Brain Sagittal Atlas if you’re working with sagittal sections. This will make it much easier to know which of your slices actually contain your ROI.
Additionally, when sectioning brains from young animals you should use an atlas of a corresponding developmental stage, such as one of the Allen Developing Mouse Brain atlases. If your developing brain atlas is not detailed enough, cross-reference with an adult brain atlas as necessary, taking into account the developmental trajectory of your ROI.
Get to know your landmarks
Keep in mind that most reference atlases you find will show tissue marked with one or more histological techniques to highlight neuron somas, myelin, nuclei, or specific markers like acetylcholinesterase. When working with live tissue you won’t have such luxuries, which means that your tissue might be significantly harder to ‘read’ than your atlas.
The main features that can readily be identified in unstained brain tissue sections are tissue edges (including hollow spaces like ventricles), major white matter areas tracts (e.g. cortical layer 1 or anterior commissure) and areas of tightly packed grey matter (e.g. CA1 area in the hippocampus). You will have to define your ROI relative to the landmarks you can actually see by eye or with a low-magnification microscope. In some areas, this will be easier to do than in others – for example, the highly ordered structure of the hippocampus or the cerebellum will be much easier to navigate than the fuzzy hypothalamic nuclei or the monotonous stripes of the striatum.

Figure 1. Bisected horizontal section of the mouse brain, demonstrating the visible contrast between grey matter and white matter tracts. Adapted from Van Hoeymissen 2020.
‘Reading’ unstained brain tissue will seem impossible at first, but you will develop an eye for it if you practice. If you’re struggling, try using a dissecting microscope or a magnifying glass. Improve your lighting with some good gooseneck illuminators or magnifier lamps. Place your tissue sections over a black background, as it will increase the contrast between grey and white matter.
Centre your objectives
When you finally place your tissue in the recording chamber, first identify your ROI using your low-magnification, 10X objective. Again, refer to the atlas and your identified landmarks to ensure that any cells you patch will definitely be in your ROI. A common approach is to position the centre of your ROI in the centre of your objective’s field of view. Then you may finally switch to your high magnification, 40X objective and start patching. A critical step in this procedure is to ensure your microscope objectives are actually centred to each other – that is, that the centre of the 40X objective field of view will actually be over the same tissue are as the centre of the 10X objective. Otherwise, you might find that you’re
always patching cells slightly to the side from your actual ROI!
A quick way to verify if your objectives are centred is to position the tip of your glass pipette exactly in the middle of the of the field of view of your 40X objective. Then, without moving your pipette, switch to your low-magnification objective. If the pipette is still exactly in the middle of the field of view, that’s perfect! Otherwise, refer to your microscope’s manual to adjust the positioning of your objectives – there is usually a dedicated screw or lever that allow for easy adjustment.
You can’t always be perfect, but you can be consistent
Hearing that there appears to be no consensus on what mouse prefrontal cortex actually is does not feel particularly great when your entire research project centres around patching mouse prefrontal cortex neurons. But this issue arises in the identification of many brain regions: defining where one area ends and another begins isn’t always easy, consistent, or uncontroversial. In addition, even in brain regions and sub-regions where a boundary is generally agreed upon (e.g. cortical layers 2 vs 3, different hypothalamic nuclei), those boundaries might not be easily identifiable in an unstained slice. How can you the be sure that you’re not accidentally patching cells in the neighbouring area instead of in your ROI?
If you cannot unambiguously label your region with the use of fluorescent markers (discussed below), a workable solution could be to clearly define your ‘safe’ cut-off criteria and adhere to them at all costs. For example, when trying to patch cells in cortical layer 2 but not layer 3, you can elect to patch only cells in the upper layer 2, and within a certain distance from layer 1 boundary. When the definition of your ROI is controversial, you can define a section of this area easily identifiable by landmarks and then report your own definition. Explicitly describe all your criteria in your Methods write-up. That way, even if your identification of your ROI is still putative, others will be able to clearly understand how it relates to other studies of this ROI.

Figure 2. Clearly and consistently define your ROI. A) diagram of a ROI (prelimbic area of the medial prefrontal cortex) in a coronal section of a mouse brain. B) Representative image of a target cell type (layer 5 pyramidal neuron), with soma made visible via Alexa Fluor 488 in intracellular solution. C) Representative cortical layer identification, with pipette to show the location of patched neuron.
Identifying your cell type
Can you make it glow?
Using fluorescent reporters is an incredibly versatile tool for identifying specific neuronal cell types. For patch clamp electrophysiology, this is frequently done using transgenic reporter lines (discussed in more depth here and here) – for example, different subpopulations of cortical interneurons can be labelled using mouse lines expressing GFP under parvalbumin (PV), somatostatin (SST), or vasoactive intestinal polypeptide (VIP). Alternatively, neurons can be fluorescently labelled using a viral vector, usually injected intracranially weeks prior to the experiment.
A cleverly planned fluorescent reporter strategy can make it remarkably easy to confidently identify your target neuronal subtype. While patching, all you might have to do is switch to a fluorescent light of the correct wavelength, take note of the cells showing sufficient fluorescence, switch back to transmitted light and proceed to patch the previously marked
cells. In addition, some patchers report that unhealthy cells tend to lose some of their fluorescence, which can help in selecting the healthiest cells for your experiment.

Figure 3. Foetal cortical plate cells transfected with Synapsin AAV.
Fluorescent reporters can be indispensable for many patch clamp experiments, but their use is not free of additional costs or caveats:
- Promoter-based fluorescent reporters can be ‘leaky’ – i.e. label neurons outside of your population of interest.
- Conversely, such systems sometimes label only a particular subset of your population of interest.
- Including genetic reporter lines can complicate your breeding scheme, increasing costs, generation time, and number of sacrificed animals.
- Viral vector-based fluorescence takes weeks to manifest, limiting its use at early developmental stages or with certain experimental timelines.
- A bright, highly expressed fluorescent marker is necessary, as the fluorescence setup at your rig will not be as high-sensitivity as a confocal microscope, and you will not be able to amplify the signal via staining.
- Certain promoters will not be robustly expressed during development, necessitating the use of less well-established cell type markers.
- There are no known markers for some neuronal subpopulations. For example, PV neurons can be divided into further subtypes, including chandelier cells and basket cells, which differ in their morphology, connectivity, function and electrophysiological properties. At the time of writing, chandelier cells cannot be easily distinguished from basket cells exclusively through the use of simple transgenic reporter lines or viral injections.
Even if your cell is labelled with a beautiful fluorescent marker, it can be terrifyingly easy to accidentally patch cells slightly below or above where you intended without even noticing – particularly in areas with tightly packed cell bodies such as the hippocampus. In such cases, it can be useful to know how to confirm the identity of your cell based on other properties, as described below. If that doesn’t reassure you, you can complete your recording as normal, but when you finish, apply a strong positive pressure to make the cell soma burst. This will make is very clear whether the cell you just recorded is your target fluorescent cell or just an inconvenient neighbour!
If it walks like a duck and quacks like a duck…
Identifying your cells based on their easily observable properties might seem old school, but in some situations it is a perfectly accurate method that can save you a lot of time and money. It works particularly well for principal neurons (i.e. cells that constitute the majority of the population in a given region) in easily identifiable ROIs, such as the hippocampus.
Properties that can be readily assessed for a patched cell are:
- Membrane resting potential
- Input resistance
- Capacitance
- Firing threshold
- Firing pattern (e.g. bursting vs regularly firing vs adapting)
- Maximum firing frequency
- Afterhyperpolarisation magnitude and shape
- Presence of inward hyperpolarisation current
- Soma size and shape
- Proximal dendrite shape and arrangement (when using a fluorescent marker like Alexa Fluor-488 in the intracellular solution)
These properties are frequently used to differentiate distinct neuron types, such as pyramidal neurons and interneurons in the cortex. Pyramidal neurons will have a larger, triangular soma, thick apical dendrite usually pointed towards layer 1, higher capacitance, lower input resistance and different firing pattern than inhibitory neurons. Inhibitory neurons can be quite diverse in their properties, but generally have a round, smaller soma (although certain types of PV interneurons have a pyramidal soma), lower capacitance and higher input resistance, variable dendritic morphology, and some show extremely fast firing rates.

Figure 4. Comparison of firing patterns shown by pyramidal neurons and two subpopulations of interneurons in rat somatosensory cortex layer 2/3. Adapted from Chistakova 2019.
Identification of neurons based on their electrophysiological properties can be extremely region- and cell-type specific, so thorough research into the cell types in your own ROI is required. In less well-characterised regions or for more fine-grained identification, sometimes it might be necessary to combine this approach with post-hoc identification, as discussed below.
Let’s double-check!
If you want even more certainty, you can add biocytin or biotin to your intracellular solution and fix the tissue section after patching. The section can then be treated with streptavidin with a fluorescent label to visualise the location and morphology of the patched neurons. You can combine this with immunohistochemistry, using antibody markers to precisely identify what types of neurons you patched post-hoc. Additional markers, for example to more clearly distinguish cortical layers, can also be used at this stage.
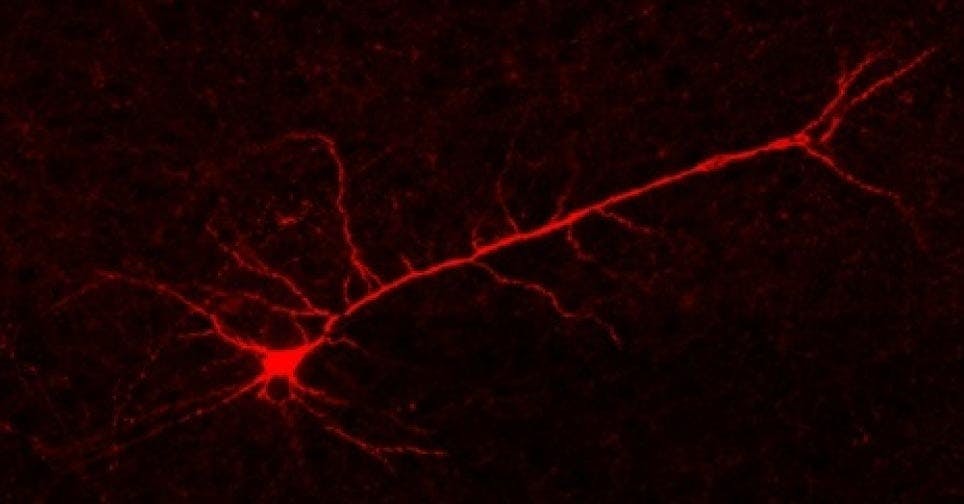
Figure 5. Biocytin-filled pyramidal neuron labelled with Streptavidin-555.
This approach can increase your workload considerably, as it necessitates extra tissue processing and imaging. But I’d personally recommend it to those new to patch clamp electrophysiology, or those who are getting used to a new model organism or brain area. When used during the initial optimisation steps of your experiment, it can be an excellent way to test if the other strategies you’re using to identify your ROI and cell types are up to the task. Additionally, it can be a way to obtain even more information about your cells, for example their morphology and dendritic spine density.
Post hoc identification methods such as biocytin-based reconstructions or more fine-grained firing pattern analysis are going to be limited if you don’t keep good documentation of your cells. In addition to the raw electrophysiological data, it might be worthwhile to record images that show the location of your cell within the whole brain slice, within your specific ROI, as well as its soma and overall visual characteristics. This way, you will always be able to go back and double-check that you’ve successfully targeted your population of interest!
References
1. Sunkin, S. M. et al. Allen Brain Atlas: an integrated spatio-temporal portal for exploring the central nervous system. Nucleic Acids Res. 41, D996–D1008 (2012).
2. Paxinos, G. & Franklin, K. The Mouse Brain in Stereotactic Coordinates. (Academic Press, 2012).
3. Van Hoeymissen, E., Philippaert, K., Vennekens, R., Vriens, J. & Held, K. Horizontal Hippocampal Slices of the Mouse Brain. J. Vis. Exp. (2020). doi:10.3791/61753-v
4. Laubach, M., Amarante, L. M., Swanson, K. & White, S. R. What, If Anything, Is Rodent Prefrontal Cortex? eneuro 5, ENEURO.0315-18.2018 (2018).
5. Li, S. et al. Overview of the reporter genes and reporter mouse models. Anim. Model. Exp. Med. 1, 29–35 (2018).
6. Abe, T. & Fujimori, T. Reporter Mouse Lines for Fluorescence Imaging. Dev. Growth Differ. 55, 390–405 (2013).
7. Haery, L. et al. Adeno-Associated Virus Technologies and Methods for Targeted Neuronal Manipulation. Front. Neuroanat. 13, (2019).
8. Chistiakova, M. et al. Distinct Heterosynaptic Plasticity in Fast Spiking and Non-Fast- Spiking Inhibitory Neurons in Rat Visual Cortex. J. Neurosci. 39, 6865–6878 (2019).
9. Swietek, B., Gupta, A., Proddutur, A. & Santhakumar, V. Immunostaining of Biocytin-filled and Processed Sections for Neurochemical Markers. J. Vis. Exp. (2016). doi:10.3791/54880
About Martyna
Martyna Panasiuk is a postdoc at the Centre of Developmental Neurobiology in King's College London investigating the effects of neurodevelopmental disorder risk genes on cortical excitation-inhibition balance using patch clamp electrophysiology, confocal imaging and RNAseq.


)